新研究利用物理信息力场,显著提升机器学习势在高温分子动力学模拟中的稳定性,为材料模拟和药物设计带来突破。
原文标题:让机器学习势活过1000K——物理学告知的原子能量模型实现前所未有的模拟稳定性
原文作者:数据派THU
冷月清谈:
怜星夜思:
2、文章强调了“先验均值函数”的重要性,它可以帮助模型在高能区域保持物理自洽。那么除了文中提到的量子化学拓扑先验,还有哪些物理或化学上的先验知识可以被引入到机器学习势中,以进一步提升模型的稳定性和准确性?
3、文章提到该模型在药物设计、新材料模拟等领域具有潜力。对于这些具体的应用场景,你认为该模型相比于传统的分子动力学模拟方法,有哪些优势和局限性?
原文内容

来源:ScienceAI本文约1500字,建议阅读5分钟从几何恢复到构象优化,全面验证物理信息力场。

机器学习势(MLP)被誉为连接量子力学精度与分子力学效率的圣杯。过去二十年,它们已经能以前所未有的精度重现能量和力的静态测试误差。
然而,一个尴尬的事实是,很多MLP在静态测试中表现完美,一旦被部署到分子动力学模拟中,尤其是在高温下,就会毫无征兆地「炸裂」——分子爆炸、键长失控、模拟崩溃。这种稳定性问题,严重制约了 MLP 在真实复杂体系中的应用。
来自英国曼彻斯特大学的团队从该痛点切入,基于「相互作用的量子原子」(IQA)框架,构建了物理学告知的高斯过程回归(GPR)原子能量模型,首次实现了在高达 1000K 的温度下、长达 10 纳秒的 NVT 模拟中「几乎无限」的稳定性。
相关研究以「Unprecedented robustness of physics-informed atomic energy models at and beyond room temperature」为题,于 2026 年 3 月 31 日发布在《Communications Chemistry》。

论文链接:https://www.nature.com/articles/s42004-026-01965-0
GPR 原子模型
本次研究中所提出的,是首个基于物理学的高斯过程(GP)原子能模型,与流行的 Behler-Parrinello 类 MLP 不同,这里报告的模型是基于预先计算且可解释的原子能训练的,其数值源自严格的量子力学定律。
为此,研究团队根据量子物理规则向模型输入了原子在现实中相互作用的详细信息,帮助人工智能对分子每个部分的运动做出更现实的预测。
图 1:基于 GP 的原子能模型的静态性能。
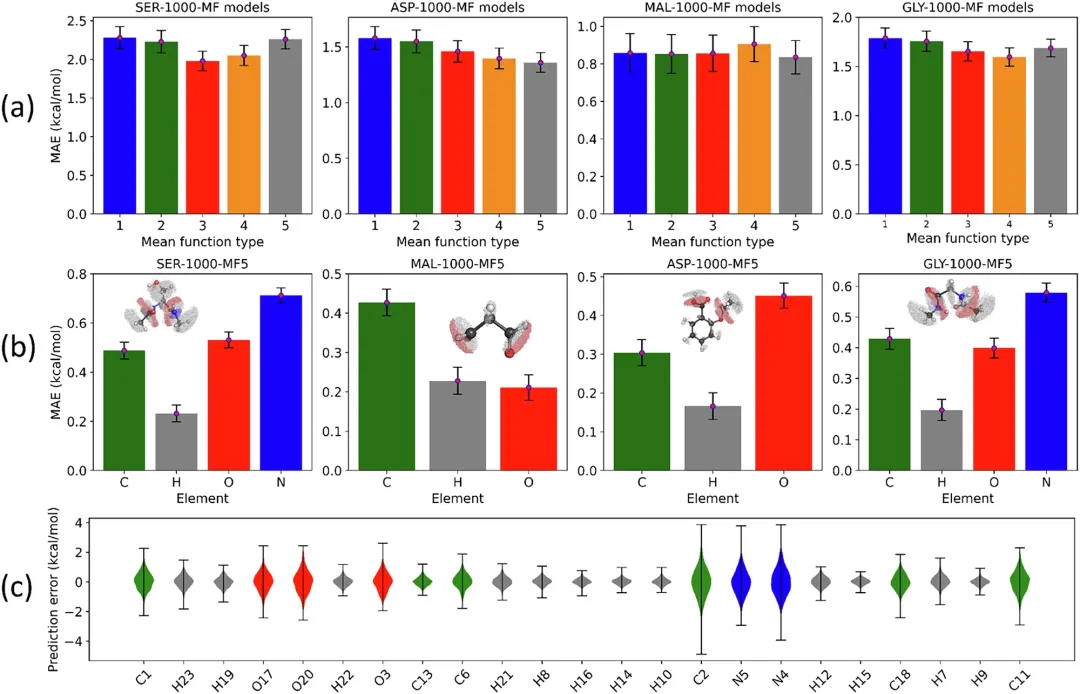
团队表示,在静态测试中,原子模型的性能不出意外地实现了相当准确的预测准确性,模型的稳健性表现也都在意料之中。
团队还发现一个小的数学选择,称为「先验均值函数」m,影响模型的稳定性;有了这个功能,人工智能就拥有了正确的「起点」,即使在分子被拉伸、加热或摇晃时,也能创建并维持稳定的模型。
图 2:基于 GP 的原子能模型的稳健性。
恢复力预测与大规模扩展模拟
在这个测试中,团队在模拟初期阶段以极不稳定的高能起始几何(SG)检查了预测的原子力,并研究了这些力如何受到模拟温度的影响。
图 3:变形 malondiardide(MAL)结构中恢复力的预测。
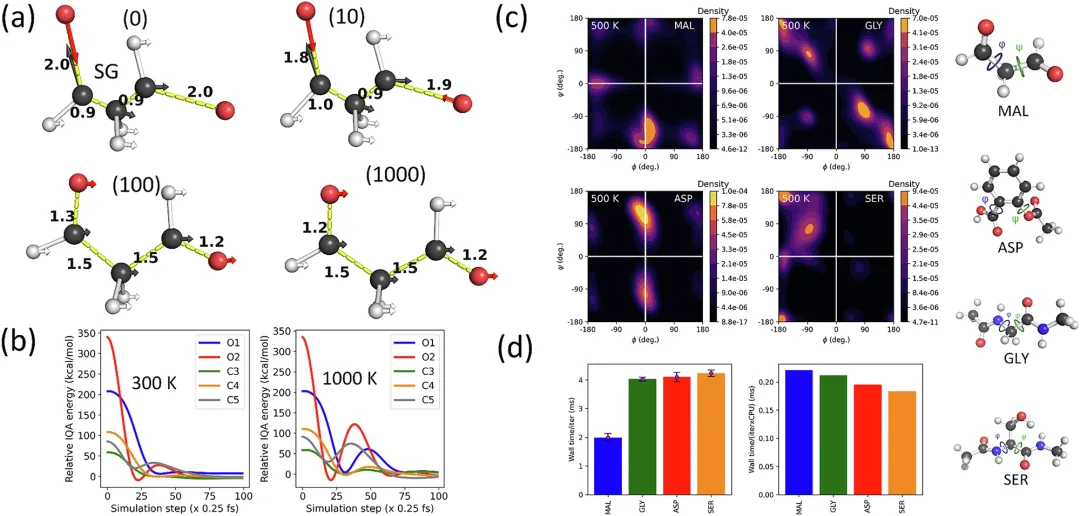
只有 MF5 模型能够成功从这些非物理结构恢复。它迅速且不可逆地松弛了非物理 SG,并在四种温度下都完成了 1 纳秒的稳定模拟。而 MF1 和 MIN 模型在几千步内即崩溃。团队分析了恢复过程中的原子力方向,发现预测的力在第一步就指向「拉伸短键、压缩长键」,这正是恢复力的直接证据。
为进一步验证长期稳定性,团队用 MF5 模型在 500K 下进行了 50 次独立的 10 纳秒模拟,总模拟时间达 0.5 微秒,全部成功完成。即使是高度柔韧的分子,如阿司匹林、丝氨酸和甘氨酸,也始终保持稳定。
构建安全可靠的分子模型
MLP 的动态稳定性不仅取决于训练数据的覆盖度,更取决于模型在外推时的「预期」。传统观念中常会假设未知区域的能量不会太高,但实际上,远离平衡态的构型能量极高。当模型低估了这些能量,就会产生错误的吸引力,导致崩溃。
对 MD 来说,模型是否稳定、是否能预测恢复力、是否能在高能区保持物理自洽,比单点 MAE 更关键。
研究团队明确指出,GP 的先验均值函数 m 决定了模型到底会不会「立刻崩掉」还是「长期运行」,而量子化学拓扑先验则给了模型一种防止任意能量波动的归纳偏差。对药物设计、新材料模拟、极端条件下的分子动力学,这种思路都意味着更可靠的长时程探索能力。
相关链接:https://www.eurekalert.org/news-releases/1121910
