Microsoft提出利用量子计算生成的数据训练AI模型,加速化学和材料科学领域的发现,实现量子精度和AI速度的结合。
原文标题:Microsoft团队提出“弯曲雅各布天梯”新思路,了解量子数据如何教会AI做更好的化学
原文作者:数据派THU
冷月清谈:
怜星夜思:
2、文中的“弯曲雅各布天梯”概念很有趣,用量子计算生成数据训练AI模型,实现了量子精度和AI速度的结合。那么,除了化学之外,这个思路在其他科学领域还有哪些潜在的应用?比如物理学、生物学等。
3、文章提到量子计算机目前仍存在错误率高和量子比特寿命有限的问题。那么,在这些问题解决之前,有没有一些“折中”的方案,可以让我们在现有技术条件下,尽可能地利用量子计算和AI的结合来加速科研?
原文内容

来源:ScienceAI本文约3500字,建议阅读5分钟量子计算机生成精确数据,AI模型学习并实现百万倍加速预测。

有时,一个视觉上引人注目的隐喻,足以让你传达一个复杂的观点。
2001 年夏天,杜兰大学物理教授 John P. Perdew 提出了一个精彩绝伦的创意。他引用圣经中“雅各布的梯子”为这个想法命名:他希望传达电子在材料中行为中固有的计算复杂性层级结构。
雅各布的阶梯代表一个梯度,佩尔杜的梯子也代表着一个梯度,不是精神层面的,而是计算层面的梯子。在最底层,数学是最简单且计算负担最小的,材料被表现为一个被简化、卡通化的原子领域。
随着不断攀升,运用越来越复杂的数学和计算能力,对原子现实的描述变得更加精确。在最顶层,自然通过极其密集的计算被完美描述——就像上帝可能看到的那样。
基于这个隐喻,或可将雅各布阶梯扩展到超越佩尔杜版本,涵盖所有模拟电子行为的计算方法。这是 Microsoft 团队设想的一种方式:它首先利用量子计算机生成关于电子行为的极其精确的数据——这些数据用经典计算成本高得令人望而却步。这些量子生成的数据随后将训练运行在经典机器上的 AI 模型,这些模型能够以惊人的速度预测材料的性质。
图示:Microsoft 的梯子想法。
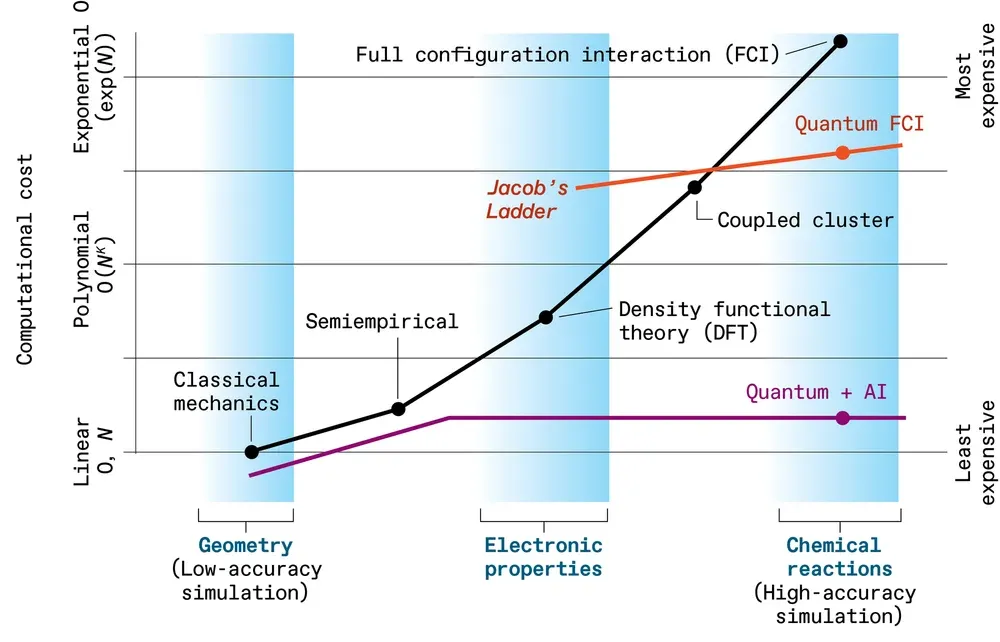
在他们的想法中,雅各布梯子的基础仍然从经典模型开始,这些模型将原子视为通过弹簧连接的简单球体——这些模型足够快,能够在长时间内处理数百万个原子,但精度最低。
而随着高度攀升,一些量子力学计算被添加到半经验方法中。最终,团队将得到单个电子的完整量子行为,但它们的相互作用将以平均化方式建模;这种更高的精度需要相当大的计算能力,这意味着你只能模拟不超过几百个原子的分子。顶端将是计算量最高的方法——在经典计算机上成本极高,但在量子计算机上则可一试。
这种量子计算与人工智能的强大结合,有望在化学发现、材料设计和复杂反应机制的理解上带来前所未有的突破。
化学和材料创新已经在我们的日常生活中扮演着至关重要——尽管常常隐形——的角色。这些发现塑造了现代世界:新药帮助更有效治疗疾病,改善健康并延长预期寿命;日常用品如牙膏、防晒霜和清洁用品;更清洁的燃料和更耐用的电池;改良化肥和农药以促进全球粮食产量等。
在这些领域,量子增强的 AI 的潜力无疑是巨大的。这些模型可以扫描此前未知的催化剂,能够固定大气中的碳,从而减缓气候变化。它们可能发现新的化学反应,将废塑料转化为有用的原材料,或者去除有毒的“永久化学物质”。
团队相信这仅仅是个开始。量子增强型人工智能将开辟材料科学的新前沿,他们也给出了几种重塑理解和控物质最根本层面能力的方法。
量子计算将如何革新化学
要理解量子计算和人工智能如何帮助打破雅各布阶梯,有必要看看目前化学中使用的经典近似技术。在原子和分子中,电子之间以复杂的方式相互作用,称为电子相关。这些相关性对于准确描述化学系统至关重要。
许多计算方法,如密度泛函理论 (DFT)或 Hartree-Fock 方法 ,通过用平均的相关性替代复杂的相关性来简化这些相互作用,假设每个电子都在由所有其他电子创造的平均场内运动。这种近似方法在很多情况下是可行的,但无法提供系统的完整描述。
电子相关在电子相互作用强烈的系统中尤为重要——例如具有特殊电子性质的材料,如高温超导体——或存在多种电子排列且能量相似的电子排列——例如含有某些金属原子的化合物,这些金属原子在催化中有着不可忽视的地位。
图示:测试中的 Microsoft 与太平洋西北国家实验室的联合项目。

在这种情况下,DFT 或 Hartree-Fock 的简化方法失效,需要更复杂的方法。随着电子配置数量的增加,计算复杂度会迅速达到“指数墙”,超过此点经典方法将变得不可行。
就在这时,量子计算机登场了。与经典比特要么开要么关闭不同,量子比特可以存在于叠加态中。这应允许它们同时表示多种电子配置,反映电子相关的复杂量子行为。由于量子计算机的工作原理与它们将模拟的电子系统相同,它们能够准确模拟即使是高度相关的系统。
人工智能在推动计算化学发展中的作用
目前,即使是计算成本较低的雅各布阶梯底层方法也很慢,而阶梯上方的方法更慢。人工智能模型已成为这类计算的强大加速器,因为它们可以作为模拟器,在不进行全部计算的情况下预测模拟结果。这些模型可以将解决问题的时间加快几个数量级。
这种加速开启了科学探索的新尺度。2023 年和 2024 年,Microsoft 与太平洋西北国家实验室 (PNNL)的研究人员合作,利用先进的人工智能模型评估了超过 3200 万种潜在电池材料,寻找更安全、更便宜且更环保的选择。用传统方法探索这一庞大的候选人群体大约需要 20 年时间。
图示:AI 模型 CDVAE 为多价电池应用生成的结构。
然而,不到一周时间, 这个名单就缩减到了 50 万种稳定材料,随后又筛选出 800 种极具潜力的候选材料。在整个评估过程中,AI 模型取代了昂贵且耗时的量子化学计算,在某些情况下,提供了比原本快达五十万倍的洞见。
随后,他们利用高性能计算(HPC)通过DFT和AI加速分子动力学模拟验证了最有前景的材料,PNNL 团队花了大约九个月时间合成并测试其中一种候选产品,并共同测试了这个产物。
这次电池突破并非独一无二。AI 模型还极大地加速了气候科学、流体力学、天体物理学、蛋白质设计以及化学和生物发现的研究。通过取代可能耗时数天甚至数周的传统模拟,AI 正在重塑跨学科科学研究的节奏和范围。
但这些 AI 模型的优劣取决于其训练数据的质量和多样性。无论是高保真模拟还是精心策划的实验结果,这些数据都必须准确反映潜在的物理现象,以确保预测的可靠性。相比之下,高质量、多样化的数据集——如那些全精度量子模拟——使模型能够跨系统推广,揭示新的科学见解。
如何加速化学发现
真正的突破将来自于战略性地结合量子计算与人工智能的独特优势。AI 已经擅长学习模式和快速预测。量子计算机仍在不断扩展以实现实用性,它们在捕捉经典计算机只能近似的电子相关性方面表现出色。所以如果你用量子生成的数据训练经典模型,你就能兼得两全其美:以 AI 的速度实现量子的准确性。
正如团队从 Microsoft-PNNL 电解质合作中了解到的,仅靠 AI 模型就能极大加快化学发现。未来,量子精确的 AI 模型将应对更大的挑战。考虑基本的发现过程,大可以把它看作一个漏斗。科学家从宽口顶部的大量候选分子或材料开始,利用滤器根据所需特性进行筛选。
量子精确的 AI 模型将显著提升化学性质预测的精度。它们能够帮助识别“首次成功”的候选分子,只将最有潜力的分子送往实验室合成和测试——这将节省时间和成本。
图示:PNNL 与 Microsoft 携手加速清洁能源解决方案的科学发现(视频片段)。

相关链接:https://youtu.be/X1aWMYukuUk
发现过程的另一个关键方面是理解控制新物质形成和行为的化学反应。可以把这些反应想象成一条蜿蜒在山地中的道路网络,每条道路代表着从起始材料到最终产品的反应步骤。反应的结果取决于它沿每条路径传播的速度,而这又取决于沿途的能量障碍。
准确度对于设计催化剂也非常重要。在这里,量子精度的 AI 模型也能发挥变革作用,提供预测反应结果和设计更好催化剂所需的高精度数据。
一旦训练完成,这些基于量子精确数据的AI模型将通过实现量子级精度,彻底革新计算化学。研究人员将能够在笔记本电脑或台式机上进行高精度仿真,而不必依赖庞大的超级计算机或未来的量子硬件。这些更容易获得的工具将使发现更加民主化,赋能更广泛的科学家群体,以应对健康、能源和可持续发展等最紧迫的挑战。
AI 与量子计算面临的挑战
到这里,许多人或许会期待变革的时刻何时会到来。
量子计算机仍面临错误率与可用量子比特寿命有限的问题,而且它们仍需扩展到有意义的化学模拟所需的规模。当前用于化学性质预测的人工智能模型可能不必完全重新设计。Microsoft 团队预计,只需先从基于经典数据预训练的模型开始,然后用量子计算机的一些结果进行微调即可。
尽管存在一些未解之谜,但 Microsoft 团队在科学理解和技术突破方面的潜在回报,使他们的提案成为该领域引人注目的方向。量子计算行业已开始超越早期噪声高的原型,十年内有望实现高精度低误差率的量子计算机。
实现量子增强人工智能在化学发现中的全部潜力,需要化学家和材料科学家(理解目标问题)、量子计算专家(负责硬件构建)以及 AI 研究人员(负责算法开发)之间的集中协作。如果做得好,量子增强的 AI 有望比任何人预期提前数年,开始应对世界上最严峻的挑战——从气候变化到疾病。
原文链接:https://spectrum.ieee.org/quantum-chemistry
