哈工大等发布DECODE,一款深度学习多组学反卷积框架,统一分析转录组、蛋白组和代谢组数据,填补代谢组学空白,助力精准医学。
原文标题:一个模型搞定各种组学!哈工大等带来通用多组学高精度反卷积框架
原文作者:数据派THU
冷月清谈:
怜星夜思:
2、DECODE框架在处理代谢组学数据反卷积方面取得了突破。代谢组学反卷积相较于转录组和蛋白组反卷积,其难点主要体现在哪些方面?
3、DECODE框架强调了在单细胞参考数据不完整的情况下,依然能准确估计已知细胞类型的相对丰度。在实际研究中,如果遇到完全没有参考数据的细胞类型,DECODE或者类似的反卷积方法还有发挥空间吗?可以尝试哪些策略来解决这个问题?
原文内容

来源:ScienceAI本文约2000字,建议阅读5分钟首次实现了对转录组、蛋白组和代谢组数据的统一、高精度反卷积,并能在参考单细胞数据不完整的情况下依然稳健工作。

在精准医学研究中,一个核心问题是:如何从大量已有的组织样本数据中,解析出其中的细胞类型组成?反卷积算法为此提供了低成本、高通量的解决方案。
不过可惜的是,现有方法大多为单一组学「量身定制」——转录组有 CIBERSORTx、蛋白组有 scpDeconv,而代谢组甚至还没有专用工具。这种「各自为战」的格局,使得跨组学、跨队列的比较充满系统性偏差,严重制约了大规模多组学研究的发展。
来自哈尔滨工业大学等的团队带来了一个名为 DECODE 的通用反卷积框架。它通过精巧的深度学习架构,首次实现了对转录组、蛋白组和代谢组数据的统一、高精度反卷积,并能在参考单细胞数据不完整的情况下依然稳健工作,为充分利用海量多组学组织数据提供了强大工具。
相关研究内容以「DECODE: deep learning-based common deconvolution framework for various omics data」为题,于 2026 年 3 月 2 日发表在《Nature Methods》。

论文链接:https://www.nature.com/articles/s41592-026-03007-y
通用反卷积框架
目前的反卷积工具遵循「针对化」发展路径。转录组工具基于特定分布假设(如泊松分布),蛋白组工具则有不同的数学模型,而代谢组反卷积仍是空白。当研究者想比较不同组学层、不同队列的细胞丰度时,方法的异质性引入了无法量化的系统偏差,破坏了整合分析的可靠性。
而在当下的组学研究中,不同组学数据在尺度、分布、稀疏性和特征维度上差异巨大。并且,单细胞参考数据往往无法覆盖组织中存在的所有细胞类型,参考数据与组织目标数据通常来自不同供体、技术和健康状态,生理和技术变异引入的批次效应会严重干扰真实生物学信号的识别。
这就要求反卷积模型架构需要高度灵活、自适应力强,且必须能在参考数据缺失某些细胞类型(即存在未知细胞类型)的情况下,依然准确估计已知细胞类型的相对丰度。这也就成为了 DECODE 的基础设计需求。
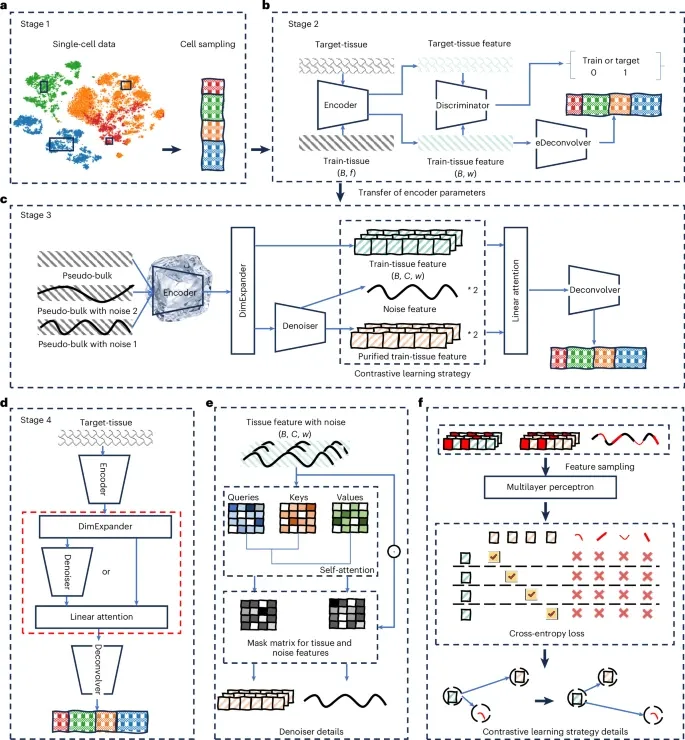
图 1:DECODE 框架。
DECODE 框架主要包含了四个阶段,这些阶段共同确保对不同组学数据的精确且可靠的解卷:
-
第一阶段:构建「伪组织」训练集:从单细胞数据中随机抽取细胞,根据随机生成的比例向量进行聚合,模拟出具有已知细胞组成的「伪组织」样本。这为模型提供了无限且带有真实标签的训练数据。
-
第二阶段:消除批次效应(对抗训练):这是 DECODE 的关键创新之一。框架引入了一个编码器、一个鉴别器和一个反卷积器。编码器提取特征,鉴别器试图判断这些特征来自伪组织还是真实目标组织,而反卷积器则专注于学习细胞组成信息。
利用对抗训练,迫使鉴别器无法区分数据来源,从而在保留生物学信号的同时,有效消除了训练数据与目标数据之间的批次效应。此阶段完成后,编码器参数被固定。
-
第三阶段:提升稳健性(对比学习与降噪):为了应对各种噪声和组学数据差异,DECODE 对每个伪组织样本随机添加不超过 10% 的噪声,构建成对训练数据。
一个注意力机制降噪器将嵌入特征分离为噪声特征和纯化特征。同时,对比学习策略拉近同一组织样本不同噪声版本的特征表示,推远不同样本的表示,从而增强模型对噪声的抵抗力。
-
第四阶段:推理与应用:DECODE 提供两条路径——当单细胞参考数据能完全覆盖目标组织细胞类型时,走「标准反卷积」路径;当存在未知细胞类型时,走带有降噪器的「相对反卷积」路径,确保依然能准确估计已知细胞类型的比例。
性能超越
研究团队设计了极其严苛的验证方案,在 15 个数据集上构建了 7 大测试场景,涵盖了转录组、蛋白组、代谢组,以及空间转录组数据。
图 2:DECODE 转录组学和蛋白质组学数据解卷积性能概述。
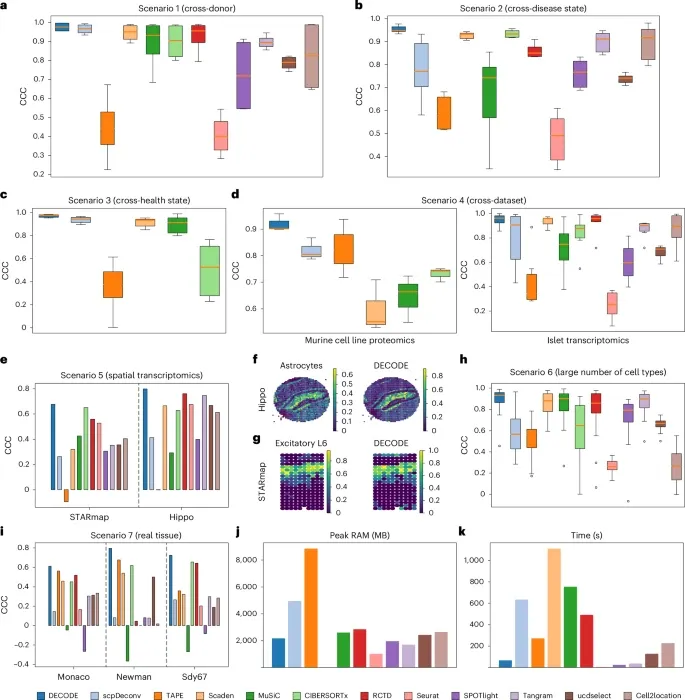
在跨供体、跨疾病状态、跨健康状态、跨数据集、空间转录组、多细胞类型等几乎所有场景中,DECODE 的一致性相关系数均排名第一。即使在部分方法的「原问题设定」下,DECODE 的表现也优于它们。在真实组织数据上,DECODE 同样展现出卓越的准确性。
图 3:DECODE 代谢组学数据解卷积性能概述。
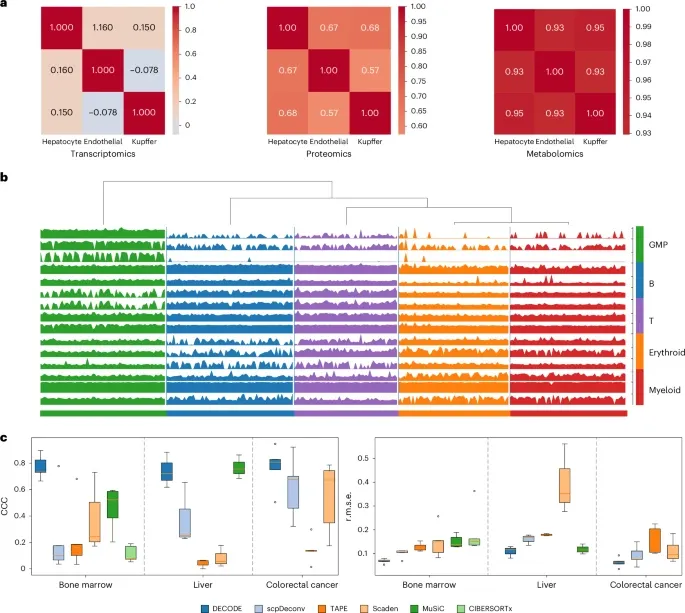
最为亮眼的是,DECODE 完成了代谢组学反卷积的突破。研究首次在三个单细胞代谢组数据集(小鼠肝脏、骨髓和人类结直肠癌)上验证了反卷积的可能性。在跨细胞类型、跨疾病状态、跨平台的测试中,DECODE 均取得了极高的预测精度。
经过四种扰动场景下的分析与在伪多组学和真实多组学队列中的应用,DECODE 在缺少模拟数据的情况下依然发表现出远超其他模型的精度。在批次效应干扰实验中,DECODE的表现也最为稳定。
通用化范式跃迁
DECODE 是一种能够处理转录组学、蛋白质组学和代谢组学数据的去卷积算法,它首次将反卷积从「组学专属」的工具箱,升级为跨组学的「通用」框架。
它不仅填补了代谢组学反卷积的空白,更重要的是,它为整合海量、异质的现有组学数据提供了统一的分析平台。有了 DECODE,研究者可以更可靠地比较不同疾病队列、不同组学层次的细胞组成变化,从而获得对复杂生物系统更完整的分子层面理解。
编辑:文婧
