华大Genos团队推出Gengram插件,通过显式基序记忆检索,提升基因组模型性能,摆脱对大规模算力的依赖。
原文标题:不用堆算力,华大新出的 Gengram,重新定义基因组建模
原文作者:数据派THU
冷月清谈:
怜星夜思:
2、Gengram的核心在于将多碱基功能基序显式建模为可检索的结构化记忆。那么,这种“记忆”的方式,除了文中提到的k-mer hash memory,还有没有其他的实现方式? 各自的优缺点是什么?
3、文章提到Gengram在motif主导任务上提升显著,尤其是在依赖短程序列模式的场景中表现突出。那么,对于非motif主导的任务,例如长程染色质互作预测,Gengram还能发挥作用吗?如果可以,应该如何改进Gengram的结构或训练方式?
原文内容

本文约3200字,建议阅读6分钟本文介绍了华大 Genos 团队推出 Gengram 轻量插件,大幅提升基因组模型性能。
华大生命科学研究院与浙江之江实验室组成的 Genos 团队在 DeepSeek 新模式的启发下,推出了一款基因组专用「外挂大脑」插件——Gengram(Genomic Engram)。仅约 2,000 万参数,即刷新了多项基因组任务的 SOTA 记录,为破解基因组建模瓶颈提供了革命性方案。
基因组基础模型(GFMs)是解码生命密码的核心工具,它们通过分析 DNA 序列解锁细胞功能、 organism 发育等关键生物信息。然而,现有基于 Transformer 的 GFMs 存在致命短板:依赖大规模预训练和密集计算间接推断多核苷酸基序,不仅效率低下,还在基序主导的功能元件检测任务中表现受限。
近日,由华大生命科学研究院与浙江之江实验室组成的 Genos 团队提出的 Gengram(Genomic Engram)模型,为这一难题提供了革命性解决方案。这一设计既避免了硬编码生物规则,又让模型获得了明确的基因组 「语法」 认知。
作为一款专为基因组基序建模设计的轻量级条件记忆模块,Gengram 的核心创新在于基于 k-mer 的 hash memory 机制,构建了可高效查询的多碱基基序记忆库。与传统模型间接推断基序不同,它直接存储 1-6 个碱基长度的 k-mer 及其嵌入向量,通过局部窗口聚合机制捕捉功能基序的局部上下文依赖,再经门控控制模块(gate-controlled module)将基序信息与主干网络融合。研究团队表示,当集成于 当前SOTA 的基因组模型 Genos 时,同等训练条件下,Gengram 在多项功能基因组学任务中实现显著性能提升,最高达 22.6%。

论文地址:https://arxiv.org/abs/2601.22203
代码地址:https://github.com/BGI-HangzhouAI/Gengram
模型权重:https://huggingface.co/BGI-HangzhouAI/Gengram
训练数据覆盖人类与非人灵长类基因组
训练数据集包含 145 个高质量的单倍型解析组装序列,涵盖人类与非人灵长类基因组。人类序列主要来源于人类泛基因组参考联盟(HPRC,第 2 版),并辅以 GRCh38 与 CHM13 参考基因组。非人灵长类序列则整合自 NCBI RefSeq 数据库,以纳入演化多样性。所有序列均使用 one hot 编码处理。词汇表包含四种标准碱基(A、T、C、G)、模糊核苷酸 N 以及文档结束标记 。
最终,系统构建了 3 套数据以支撑消融实验及正式预训练
50B tokens @ 8,192(消融)
200B tokens @ 8k(10B 正式预训)
100B tokens @ 32k(10B 正式预训)
并且保持 human : non-human = 1:1 的数据混合比例。
基因组建模从「注意力推导」走向「记忆增强」
受 DeepSeek Engram 记忆机制启发,Genos 团队快速开发并部署 Gengram,为基因组基础模型提供显式 motif 存取与复用能力,突破主流 GFMs 缺乏结构化 motif memory、只能通过扩大训练数据「隐式记忆」的限制,推动基因组建模从「注意力推导」走向「记忆增强」。该模块架构如下图所示:
Gengram 架构图
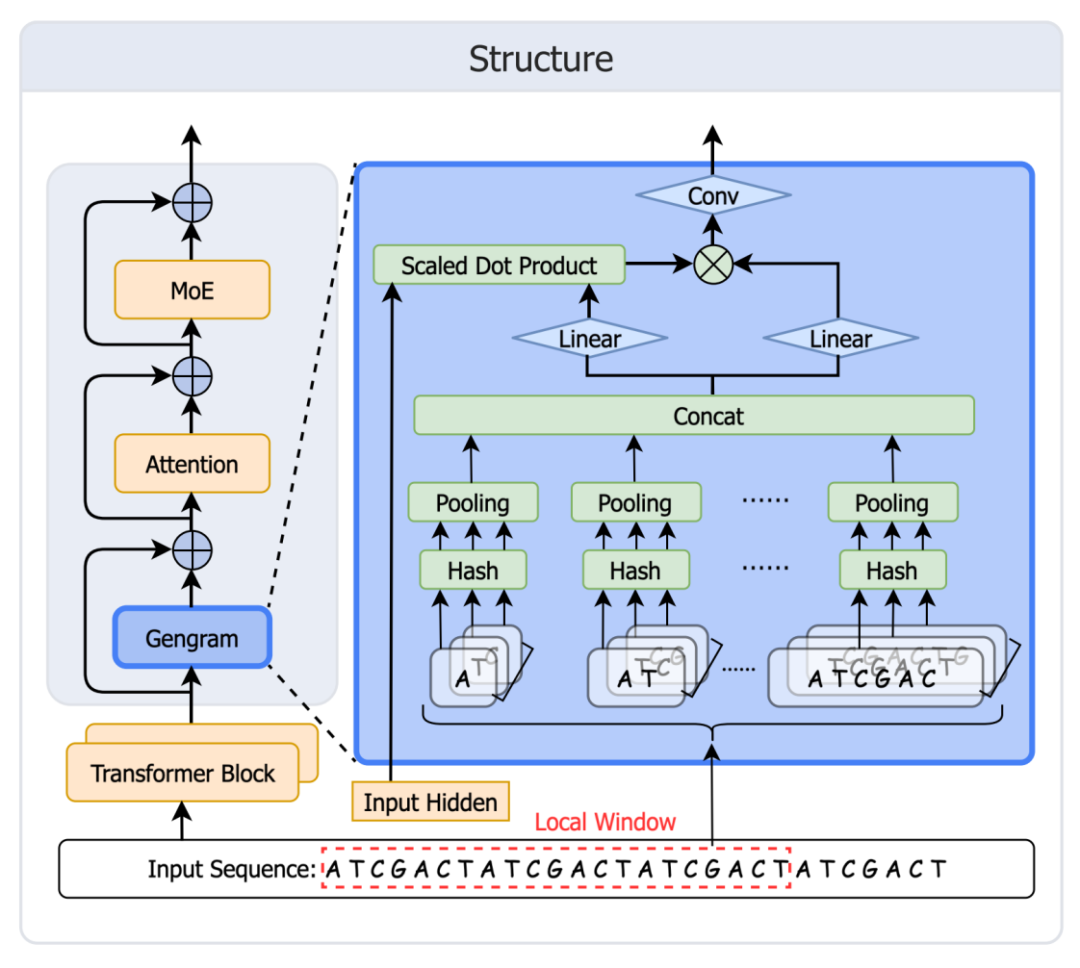
建表:对 k=1~6 的所有 k-mer 建立 hash memory(静态 key + 可学习 embedding value);
检索:把窗口内出现的所有 k-mer 映射到表项;
聚合:先在每个 k 上聚合,再跨 k 拼接;
门控:gate 控制激活,把 motif 证据写入 residual stream,然后再进入 attention。
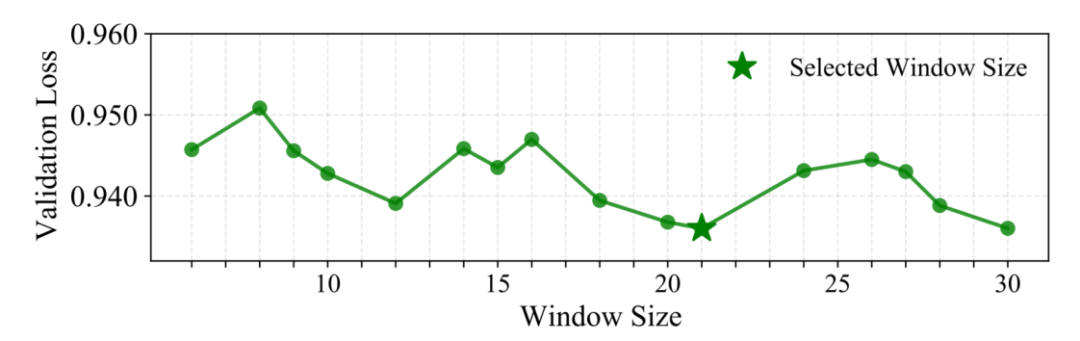
一个关键设计:Local Window Aggregation(W=21bp)
Gengram 并非在每个位置仅检索单一 n-gram,而是采用固定窗口内的多 k-mer embedding 聚合,以更稳定地注入「局部、结构一致」的 motif 证据。研究人员通过窗口大小策略搜索进行验证,发现 21 bp 在验证集上达到最优性能。一个可能的生物学解释是:典型的 DNA 双螺旋周期约为每旋转一圈 10.5 个碱基对,因此 21 个碱基对正好旋转两圈;这意味着,相隔 21bp 的两个碱基,在三维空间中恰好位于螺旋的同一侧,面对相似的生化环境,在该尺度上进行窗口聚合,或更有利于对齐局部序列信号的相位一致性。
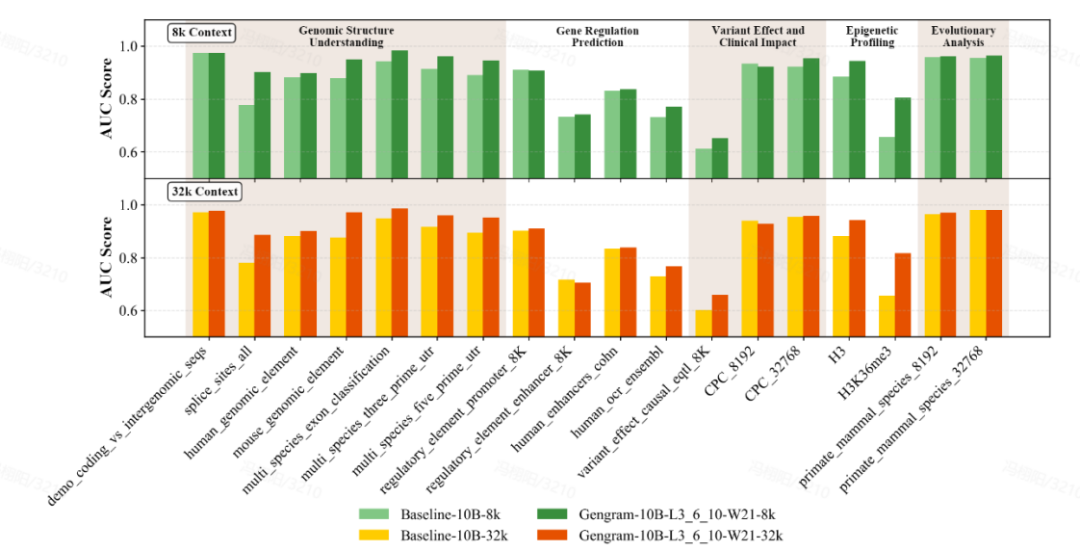
评测提升突出:小参数,大改变
团队采用多标准基准数据集对模型进行了全面评估,涵盖 Genomic Benchmarks (GB)、Nucleotide Transformer Benchmarks (NTB)、Long-Range Benchmarks (LRB)及Genos Benchmarks (GeB)。从中选取了 18 个具有代表性的数据集,涉及 5 个主要任务类别:序列结构理解 (Genomic Structure Understanding)、基因调控预测 (Gene Regulation Prediction)、表观遗传图谱 (Epigenetic Profiling)、变异效应与临床影响 (Variant Effect & Clinical Impact) 以及进化分析 (Evolutionary Analysis)。
Gengram 作为一个仅约 2,000 万参数的轻量化插件,相对于百亿级规模的基座模型而言参数占比极小,但其带来的性能提升显著。在 8k 与 32k 两种上下文长度设定下,同等训练条件,集成 Gengram 的模型在绝大多数任务中均优于未集成的版本。具体表现上,剪接位点预测任务的 AUC Score 从 0.776 提升至 0.901,增幅达 16.1%;表观遗传预测任务(H3K36me3)的 AUC Score 从 0.656 提升至 0.804,增幅为 22.6%。
8k 和 32k context 下,加入 Gengram 前后的评测结果,加入 Gengram 后提升显著
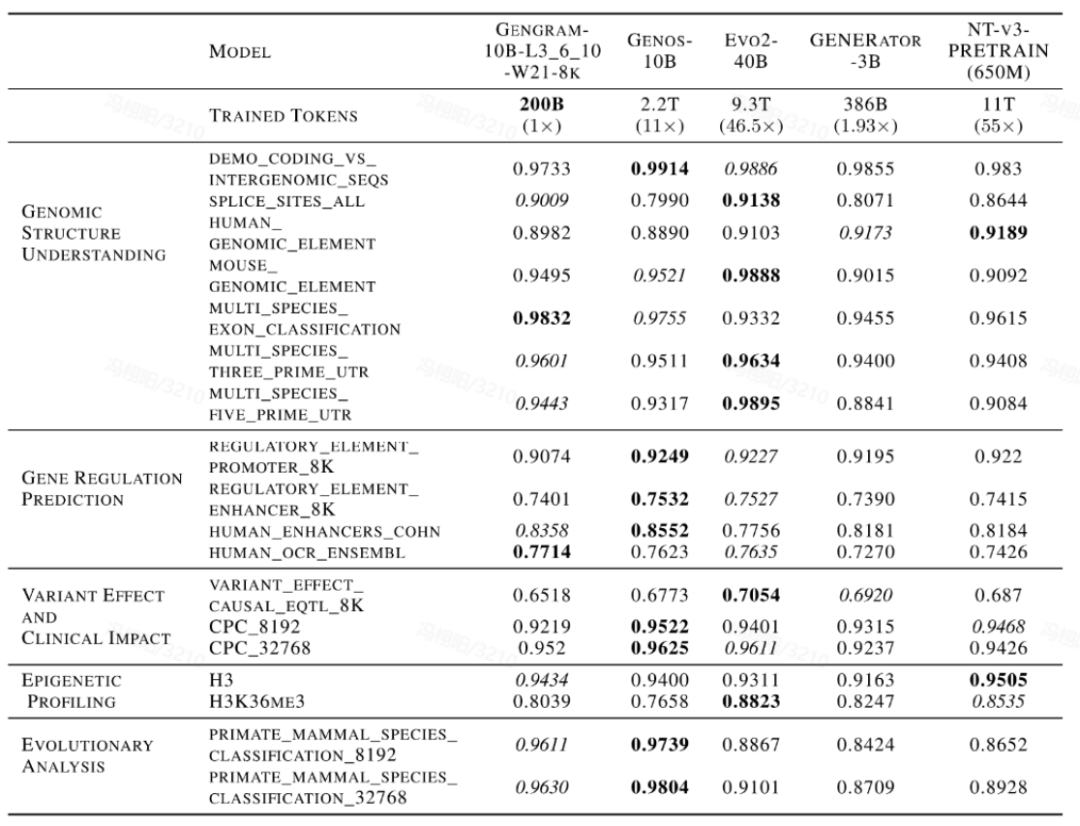
此外,该性能提升还伴随着显著的「数据杠杆」效应。在与 Evo2、NTv3、GENERATOR-3B 等主流 DNA 基础模型的横向对比中,集成 Gengram 的模型仅需极小规模的训练数据和较少的激活参数量,便可在核心任务上媲美训练数据规模领先其数倍至数十倍的公开模型,体现出较高的数据训练效率。
Gengram 模型也主流 DNA 大语言基础模型的评测比较
深度剖析 Gengram
为什么 Gengram 能加速训练?
团队引入 KL 散度作为训练过程的表征诊断指标,并采用 LogitLens-KL 对不同层的「可预测性(prediction-readiness)」进行量化跟踪。结果显示,引入 Gengram 后,模型在浅层即可更早形成稳定的预测分布:相较基线模型,其层间 KL 更快下降并提前进入低值区间,表明有效监督信号更早被组织为可用表征,从而使梯度更新更直接、优化路径更平滑,最终体现为更快的收敛速度与更高的训练效率。
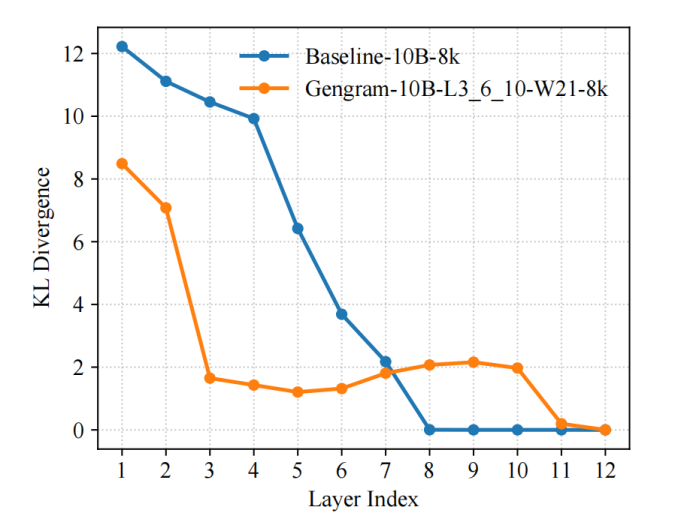
这一现象并非「凭空发生」,而是由 Gengram 的结构性设计直接驱动:
显式的 motif 记忆检索,缩短「证据到表征」的路径。 在基因组任务中,监督信号往往由短而稀疏的 motif(如剪接共识序列、启动子相关片段、低复杂度 tract 等)触发。基线 Transformer 需要通过多层 attention/MLP 逐步「推导并固化」这些局部证据;而 Gengram 通过对 k-mer 的显式存取,把这类高信息密度的局部模式以记忆形式直接提供给网络,使模型不必等待深层逐渐形成 motif detectors,从一开始就更接近可预测状态。
窗口聚合 + 动态门控,使注入的证据「稳定且可控」。 Gengram 不是逐位置硬注入,而是在固定窗口内聚合多个 k-mer embedding,并通过门控选择性写入 residual stream:在功能区域更倾向激活检索,在大段背景区抑制检索。这种「稀疏、对齐功能元件」的写入方式,一方面减少噪声干扰,另一方面让网络更早获得高信噪比的训练信号,降低了优化难度。
Motif 记忆从何而来?详解 Gengram 的写入机制
研究团队在下游评测中首先观察到一个明确且跨任务一致的现象:在相同训练设定下,引入 Gengram 后,模型在典型的 motif 主导任务上取得显著提升,尤其是在依赖短程序列模式的场景中表现突出,例如剪切位点识别与表观遗传相关的组蛋白修饰位点预测。以代表性任务为例,剪接位点预测 AUC 从 0.776 提升至 0.901,H3K36me3 预测 AUC 从 0.656 提升至 0.804,增益稳定且幅度可观。
为了进一步回答「这些提升从何而来」,团队没有止步于指标层面,而是从模型前向传播中提取 Gengram 的残差写入项(residual write),并将其在序列维度上的强度分布可视化为热图进行分析。结果显示,写入信号呈现出高度稀疏且强对比的结构:绝大多数位置接近基线,只有少数位置形成尖锐峰值;更重要的是,这些峰值并非随机出现,而是显著富集并对齐于功能相关区域与边界,包括启动子邻近的 TATA-box 片段、低复杂度 poly-T 片段,以及基因/外显子等功能区域边界附近的关键位置。这意味着 Gengram 的写入更像是在「抓住决定功能的局部证据」,而非无差别地在全序列范围内注入信息。
综合上述现象与证据链,研究人员可以将 Gengram 的 motif 记忆机制概括为「按需检索—选择性写入—结构化对齐」:模块通过门控控制检索与写入强度,在功能信息密度更高的区域更积极地注入可复用的 motif 证据,在背景区域则抑制写入以降低噪声干扰。由此,模型对 motif 的掌握不再主要依赖更大规模数据带来的「隐式记忆」,而是转向一种显式存取、可解释地写入表征的结构化能力。

结语
近年来,基因组建模领域正经历从「序列统计学习」向「结构感知建模」的关键转向。
以 Gengram 为代表的条件化基序记忆机制,揭示了一条不同于传统密集计算的技术路径:通过将多碱基功能基序显式建模为可检索的结构化记忆,模型得以在保持通用架构兼容性的同时,实现更高效、更稳定的功能信息利用。这一思路不仅在多项功能基因组任务中展现出显著性能优势,也为稀疏计算、长序列建模以及模型可解释性提供了统一的工程解法。
此外,从产业视角看,Gengram 所体现的「结构化先验 + 模块化增强」范式,显著降低了基因组大模型在算力、数据与训练周期上的边际成本,为其在药物研发、变异筛选、基因调控分析等高价值场景中的规模化部署提供了现实可行性。更长远地看,这类可复用、可插拔式的架构组件,或将成为下一代基因组基础模型的标准配置,推动行业从「更大的模型」走向「更聪明的模型」,并加速学术研究成果向产业平台与临床应用的持续转化。
作者:Genos Team
