SUICA模型结合INR与图自编码器,精准预测空间转录组基因表达,提升数据质量,克服零膨胀与高成本难题。
原文标题:数据降噪/生物信号强化/缓解dropout,深度学习模型SUICA实现空间转录组切片中任一位置基因表达的预测
原文作者:数据派THU
冷月清谈:
怜星夜思:
2、文章里提到空间转录组能“把‘表达了哪些基因’与‘位于组织的哪一处’绑定在一起”,这和传统基因测序只知道基因表达量,但不知道位置,相比起来,对疾病诊断或者新药研发具体能带来哪些革命性的变化啊?感觉这个应用场景挺大的!
3、SUICA模型用了“隐式神经表征”和“图自编码器”这两种技术,看起来挺酷的。除了空间转录组,大家觉得这俩技术组合起来,还能应用到哪些别的生物医学大数据分析领域啊?比如蛋白质结构预测、药物分子筛选之类的行不行?
原文内容

来源:HyperAI 超神经
本文约2600字,建议阅读9分钟
东京大学郑银强老师组,麦吉尔大学丁俊老师组共同提出了一种针对空间转录组数据建模的方法 SUICA。
东京大学郑银强老师组,麦吉尔大学丁俊老师组共同提出了一种针对空间转录组数据建模的方法 SUICA。

SUICA 是一个基于隐式神经表征(implicit neural representations, INR)和图自编码器(Graph-Autoencoder)的深度学习模型。SUICA 使用图自编码器对高维的空间转录组数据进行降维,然后使用隐式神经表征对空间转录组数据坐标和其对应的基因表达进行建模,从而实现空间转录组切片中任一位置基因表达的预测。结果证明,通过 SUICA 处理的空间转录组数据能够有更高的质量,更低的噪声和更强的生物信号。
相关成果以「SUICA: Learning Super-high Dimensional Sparse Implicit Neural Representations for Spatial Transcriptomics」为题,入选 ICML 2025。

论文地址:
https://go.hyper.ai/C6Zcl
更多 AI 前沿论文:
https://go.hyper.ai/owxf6
什么是空间转录组数据?
空间转录组(Spatial Transcriptomics, ST)数据是在同一张组织切片上同步记录「基因表达量」和「空间坐标」的高维信息矩阵。与传统的只能呈现形态学结构的全景组织影像(WSI)或只能量化基因表达但丢失方位的常规转录组测序(Transcriptomics)相比,空间转录组把「表达了哪些基因」与「位于组织的哪一处」绑定在一起,绘制出组织内细胞状态与微环境相互作用的功能地图,因而成为连接组织学和分子组学的新型数据形态。
为什么需要增强空间转录组数据?
尽管空间转录组带来了前所未有的空间分辨分子视角,但现实数据仍受到三大瓶颈限制:
① 分辨率–成本矛盾:探针越密、测序深度越高,实验费用(如 stereo-seq 的测序实验成本大于$4,000 /cm²)和样本通量迅速攀升;
② 信号稀疏与噪声:每个探测点捕获的 mRNA 数量有限,零膨胀严重,导致低丰度或关键调控基因易被漏检;
③ 跨平台异质性:不同平台在探针物理排布、测序深度和背景噪声上差异显著,直接阻碍多样本或多实验整合。
计算增强方法包括超分辨重建、深度去噪与缺失值填补等,可以在不增加(或仅小幅增加)实验成本的前提下:
(a) 预测未测序到点位的基因表达;
(b) 恢复因为技术限制未能检测到的真实基因表达,提升差异基因与空间可变基因检测灵敏度;
(c) 生成在不同平台之间可比、可共享的标准化特征表征。
由此为细胞通讯解析、疾病分区注释、药物靶点发现、多组学联合建模和 AI 病理辅助诊断提供更加精准、丰富且可扩展的数据基础,极大释放空间转录组技术在基础研究和临床转化中的潜力。
SUICA:基于隐式神经表征和图自编码器的统一模型
利用隐式神经表征对空间转录组数据建模的挑战
空间转录组数据的建模面临多重挑战:
首先,原始数据在空间维度上呈网格状分布,而在基因维度上则高达数千到上万,形成「超高维、极稀疏、噪声大」的矩阵;高 dropout 率导致生物学关键信号被弱化,加剧了统计功效不足。
其次,现有空间转录组平台在「分辨率—成本」之间存在根本权衡——探针越密、测序越深,成本便成倍攀升,因而难以同时获得细胞级分辨率和大规模样本量。
再次,尝试用隐式神经表示将离散空间转录组点插值为连续表达场时,需同时解决两大技术难点:一是基因表达空间的维度远超传统视觉信号,单纯扩宽或加深网络难以摆脱维度灾难;二是零膨胀导致输入信号分布高度不均,常规 INRs 难以捕捉复杂且非线性的空间表达模式。
图自编码器:将高维空间转录组数据降维
相较于传统自编码器,我们先把每个空间转录组中的数据点视为图节点、以空间邻近关系构建邻接矩阵,然后在编码器中采用图卷积对原始高维基因表达进行卷积,将局部空间上下文融入表示并压缩到低维表征;以此学习高维空间转录组数据的低维表征,而图卷积的加入可以加强稀疏、大噪声的空间转录组数据信号。
隐式神经表征:建立测序点坐标与基因表达间的映射
在获得低维表征后,隐式神经表征网络接收检测点的坐标为输入,学习「点」与其对应低维表征的映射。并且将学习后的,模型预测的低维表征送入图自编码器中的解码器部分,从而实现将坐标映射到高维基因表达的作用。
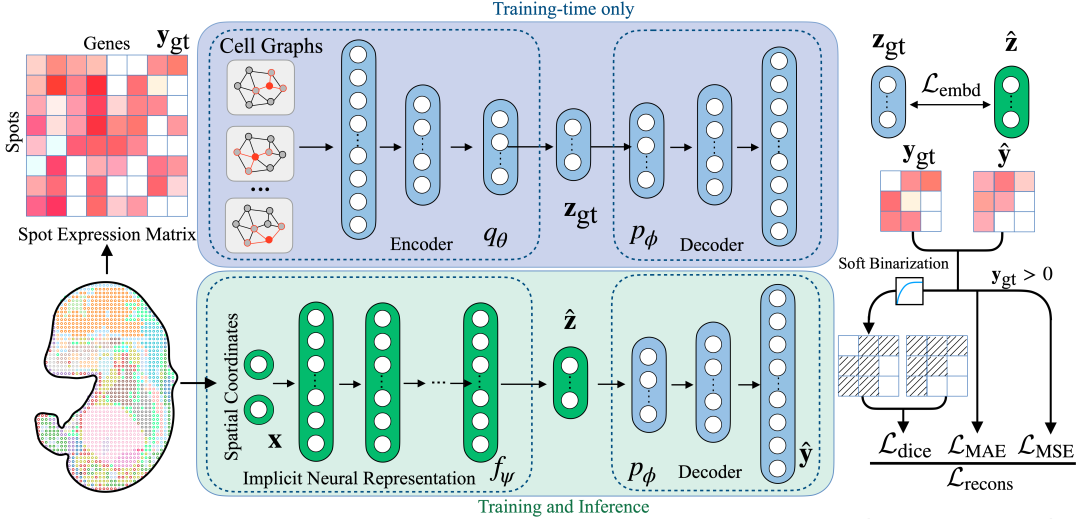
SUICA 模型架构图
实验验证:SUICA 能生成更精准和具有更强生物相关性的预测结果
我们利用 stereo-seq 的老鼠胚胎数据和 Slide-seq 老鼠脑部切片数据进行基准对比,在未知点位预测(超分辨率)的任务上 SUICA 在多个关键指标上显著优于现有的模型和传统的隐式神经表征模型,包括 FFN,SIREN。我们可视化了每种方法的预测效果,结果显示 SUICA 的预测不仅能够准确的复原出基因的表达模式,甚至能够增强基因的表达信号。如 SEPT3,该基因在老鼠胚胎的神经系统发育中扮演着重要的作用,尽管在 groundtruth 中的信号并不明显,但 SUICA 的预测结果成功地捕捉到了这一信号。
通过对各种方法生成的结果进行聚类与标注,我们直观地发现 SUICA 生成的细胞类型最接近于真实的细胞类型。并且 SUICA 生成的细胞类型中在空间上保留了更为细节的器官和组织结构。这些结果说明 SUICA 有能力增强生物信号,并且能够识别不同器官、组织之间的细微细胞状态区别。
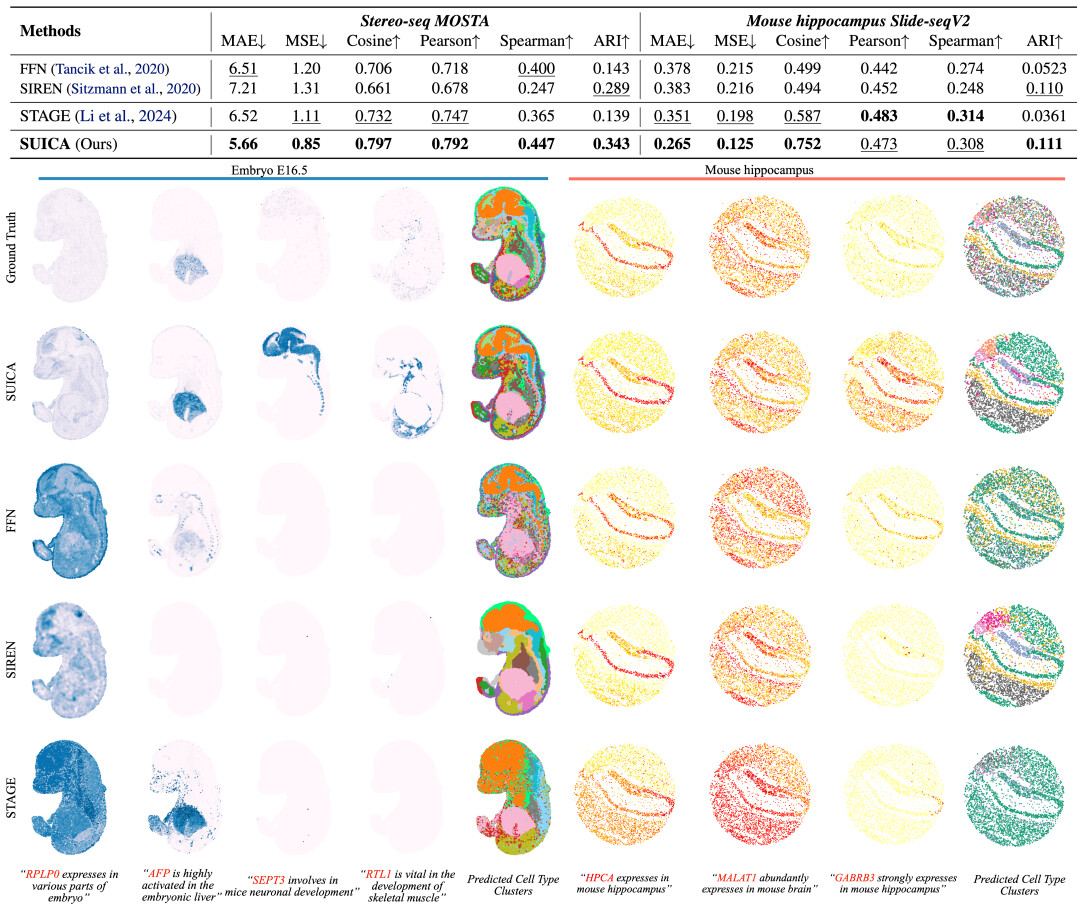
SUICA 生成细胞实验数据
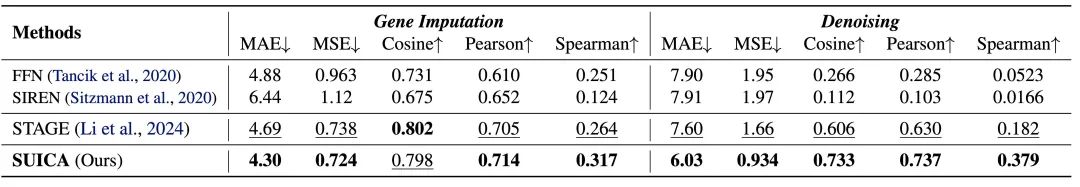
实验验证:SUICA 能够减少空间转录组数据的噪声并且缓解 dropout 现象
为了验证 SUICA 的去噪能力(denosing)和其从 dropout (因为测序技术限制导致的读数为 0 的结果)中恢复真实基因表达的能力(gene imputation),我们人为地对空间转录组数据加入高斯噪声或随机将基因表达设为 0。在 gene imputation 实验中,我们随机的将数据中百分之 70 的基因表达设为 0。 在基因表达在去噪实验中,为了保证加噪后的基因表达分布仍然与原始基因表达分布相似,我们将所有的负值归零。实验结果表明 SUICA 在多项指标上强于现有的方法,证明了 SUICA 能对空间转录组数据降噪并且缓解 dropout 现象的能力。
编辑:王菁
