浙江大学团队提出SIMO算法,利用概率对齐整合多组学单细胞数据,实现空间重构和基因调控分析,为疾病研究提供新视角。
原文标题:Nat. Commun. | 基于概率对齐的多组学数据空间整合方法
原文作者:数据派THU
冷月清谈:
浙江大学范骁辉、陆晓燕团队在《Nature Communications》上发表文章,介绍了一种名为SIMO(Spatial Integration of Multi-Omics)的新算法,旨在解决多模态单细胞数据空间整合的难题。SIMO通过概率对齐和最优传输算法,能够整合空间转录组数据以及其他单细胞多组学数据,重构细胞类型的空间分布,并揭示基因调控的空间模式。该方法采用分步对齐策略,首先整合空间转录组和单细胞转录组数据,然后整合非转录组数据,如染色质可及性数据。SIMO在模拟和真实数据集上均表现出优于现有工具的性能,尤其在处理非转录组数据和多模态数据时优势明显。研究结果表明,SIMO能够准确重构多组学细胞类型的空间分布,揭示不同组学之间的生物学关系,并能应用于小鼠胚胎、大脑以及人类心肌梗死等生物数据集的分析,为深入理解组织生理和病理状态提供重要依据。该工具为疾病研究和治疗策略开发提供了新的视角。
怜星夜思:
1、SIMO算法中,超参数α如何影响整合结果?实际应用中如何选择合适的α值?
2、SIMO算法在处理不同类型的组学数据时,如何保证整合的准确性和生物学意义?
3、SIMO算法在疾病研究和治疗策略开发方面有哪些潜在应用?除了文中提到的小鼠皮层和人类心肌梗死,还可以应用在哪些疾病或研究方向?
2、SIMO算法在处理不同类型的组学数据时,如何保证整合的准确性和生物学意义?
3、SIMO算法在疾病研究和治疗策略开发方面有哪些潜在应用?除了文中提到的小鼠皮层和人类心肌梗死,还可以应用在哪些疾病或研究方向?
原文内容

来源:IntelliOmics本文约2000字,建议阅读5分钟
随着单细胞组学测序技术的发展,SIMO有望更全面地整合关键基因调控数据,在疾病研究和治疗策略开发等方面具有巨大潜力。
论文背景
本文由浙江大学范骁辉、陆晓燕团队发表于Nature Communications期刊。文章链接附在本文文末。

空间组学技术和单细胞组学技术的飞速发展为解析组织微环境的分子机制提供了高分辨率工具。然而,现有的空间组学技术主要聚焦单一模态(如转录组或蛋白质组),难以同时捕获染色质可及性、DNA甲基化等多模态信息,限制了在多模态水平上全面解析组织生物学的能力;单细胞测序技术虽然能提供多种组学数据,但组织解离导致了空间信息的丢失,难以在空间背景下进行综合分析。
现有的计算方法主要集中在整合空间转录组数据与单细胞RNA测序数据,或在已配对的多组学数据上进行空间映射;然而,这些方法要么只关注转录组学,要么依赖于配对数据,要么不能有效地纳入空间信息。因此,开发能够整合多种单细胞多组学数据并保留其空间信息的工具,对于深入理解组织的空间生物学至关重要。
模型构建
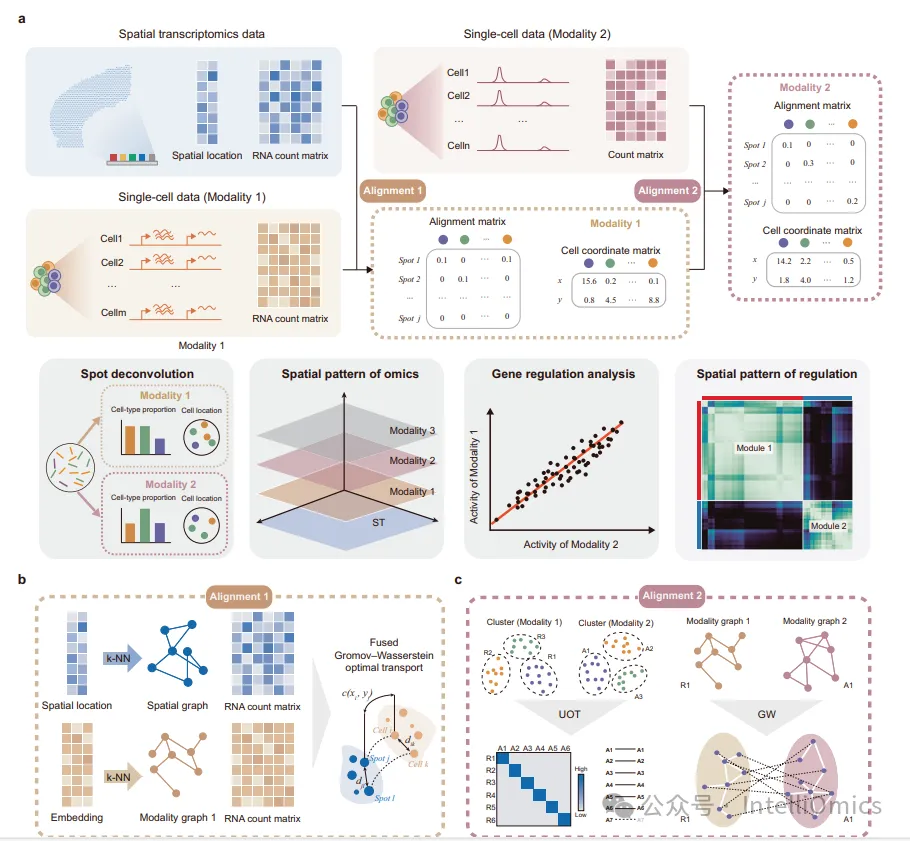
文章提出了SIMO(Spatial Integration of Multi-Omics)算法,通过概率对齐与最优传输算法,实现多模态单细胞数据的空间整合。
SIMO采用分步对齐策略,依次整合不同模态数据。首先,SIMO基于空间转录组数据和转录组数据源于同一模态的前提进行整合,借鉴了先前的开发工具SpaTrio的计算策略,利用kNN算法构建空间图(基于空间坐标)和模态图(基于低维嵌入),并通过融合的Gromov-Wasserstein最优传输计算细胞与斑点(组织切片被划分为众多微小的区域)之间的映射概率矩阵。在计算过程中,设置关键超参数α,用于平衡基因表达相似性与图结构相似性:α的取值范围在0到1之间,较小的α值使整合更侧重基因表达相似性,更关注细胞和斑点在转录组层面的相似程度;较大的α值则更偏向图结构相似性,强调细胞和斑点在空间图和模态图中的相对位置和连接关系。最后基于映射细胞与其周围斑点的转录组相似性微调细胞坐标。
整合转录组数据和非转录组数据时,SIMO对scRNA-seq数据和另一模态的数据(以scATAC-seq数据为例)进行预处理,得到相应模态的低维表示并构建kNN图,使用Leiden算法为初始细胞簇分配标签。模型以基因活性分数作为连接RNA和ATAC模态的“桥梁”,计算不同模态细胞簇的基因表达(mRNA表达量)与基因活性分数(基因启动子及增强子区域的开放性程度)的平均皮尔逊相关系数(Pearson Correlation Coefficients,PCCs),利用非平衡最优传输(Unbalanced Optimal Transport,UOT)算法促进模态间的标签转移。接着,为具有相同标签的细胞组构建特定模态的kNN图并计算距离矩阵,通过Gromov-Wasserstein传输计算确定不同模态数据集之间细胞的对齐概率,最后根据细胞匹配关系将scATAC-seq数据精确分配到特定空间位置,并基于低维嵌入表示和余弦相似性来衡量细胞与周围斑点的关系,进而调整坐标。通过修改UOT成本矩阵的构建方法,SIMO可实现各种组学类型的空间映射。
下游分析方面,基因调控分析根据具体分析需求,将数据转化为以基因名称为特征的矩阵,如从ATAC数据计算的基序活动矩阵。通过计算基序活动的倍数变化与基因表达之间的PCCs,分析不同细胞群体之间的相关性和调控模式。空间调控分析整合两种模态的数据及其空间信息,应用空间平滑算法减少数据噪声,并使用跨模态平滑补充模态之间的信息,计算跨模态的基因对的表达比例作为调控分数,以评估基因调控强度。基于空间位置信息构建核矩阵,通过加权相关分析和共识聚类识别具有相似空间调控模式的特征模块。
研究结果
研究结果显示:在模拟数据集评估中,SIMO在不同复杂程度和噪声水平下都展现出高准确性和稳定性,超参数α设为0.1时性能最佳;与CARD、Tangram等现有工具对比,SIMO在模拟和真实数据集上均表现更优,尤其在处理非转录组数据和多模态数据时优势明显;在生物数据评估方面,对小鼠胚胎、小鼠大脑等不同生物数据集的分析,证明了SIMO能准确重构多组学细胞类型的空间分布和各种组学特征,揭示不同组学之间的生物学关系;应用于小鼠皮层和人类心肌梗死的空间整合研究时,SIMO不仅能呈现细胞类型的空间分布和基因调控机制,还挖掘出潜在的治疗靶点,为深入理解组织生理和病理状态提供了重要依据。
讨论
SIMO是一种利用概率对齐和最优传输算法,通过顺序空间映射来整合多模态单细胞组学数据的计算工具,具备强大的下游分析能力。与现有方法相比,SIMO优势显著,能同时重构多种模态数据的空间分布,深入探究基因调控的空间模式,且理论上可兼容各类与转录组相关的组学数据。随着单细胞组学测序技术的发展,SIMO有望更全面地整合关键基因调控数据,在疾病研究和治疗策略开发等方面具有巨大潜力。
原文链接:
https://doi.org/10.1038/s41467-025-56523-4
参考文献
[1] Yang P, Jin K, Yao Y, Jin L, Shao X, Li C, Lu X, Fan X. Spatial integration of multi-omics single-cell data with SIMO. Nat Commun. 2025 Feb 1;16(1):1265.
编辑:文婧
